Support for ClinicalTrials.gov Compliance
ClinicalTrials.gov is a database of privately and publicly funded clinical studies providing information about clinical trials to patients, family members, health care professionals, researchers and the larger public. Investigators are responsible for compliance with all regulations related to public disclosure of clinical trial information.
Failure to comply with this policy can lead to financial penalties, withholding of federal grant money or requirement to pay money back, and/or inability to publish results. It is Ohio State policy that clinical trials must be registered and summary results reported on ClinicalTrials.gov in accordance with federal regulations and NIH policy.
This site hosts information for Administrators and Investigators to help support compliance with ClinicalTrials.gov regulations.
The ClinicalTrials.gov Protocol Registration System is the portal used to manage clinical trial information.
Information for Administrators
This document provides a summary of responsibilities of an organization's ClinicalTrials.gov Protocol Registration and Results System (PRS) Administrator.
Administrators have three primary responsibilities:
- Maintain their organization's PRS account, including monitoring for problems with records
- Approve and release study records and updates when the Sponsor is the Responsible Party
- Serve as the primary point of contact with the ClinicalTrials.gov team
Each college will have at least one designated CT.gov PRS administrator that will be the point person for the college. Additional administrators can be added if the level of work warrants additional administrators.
Administrator accounts can be created by the college admin or requested from Sandra Meadows.
Logging into the System
Protocol Registration and Results System Login is not found on ClinicalTrials.gov, the login is on the registration page.
Responsibilities of the Administrator
Create user/administrator accounts.
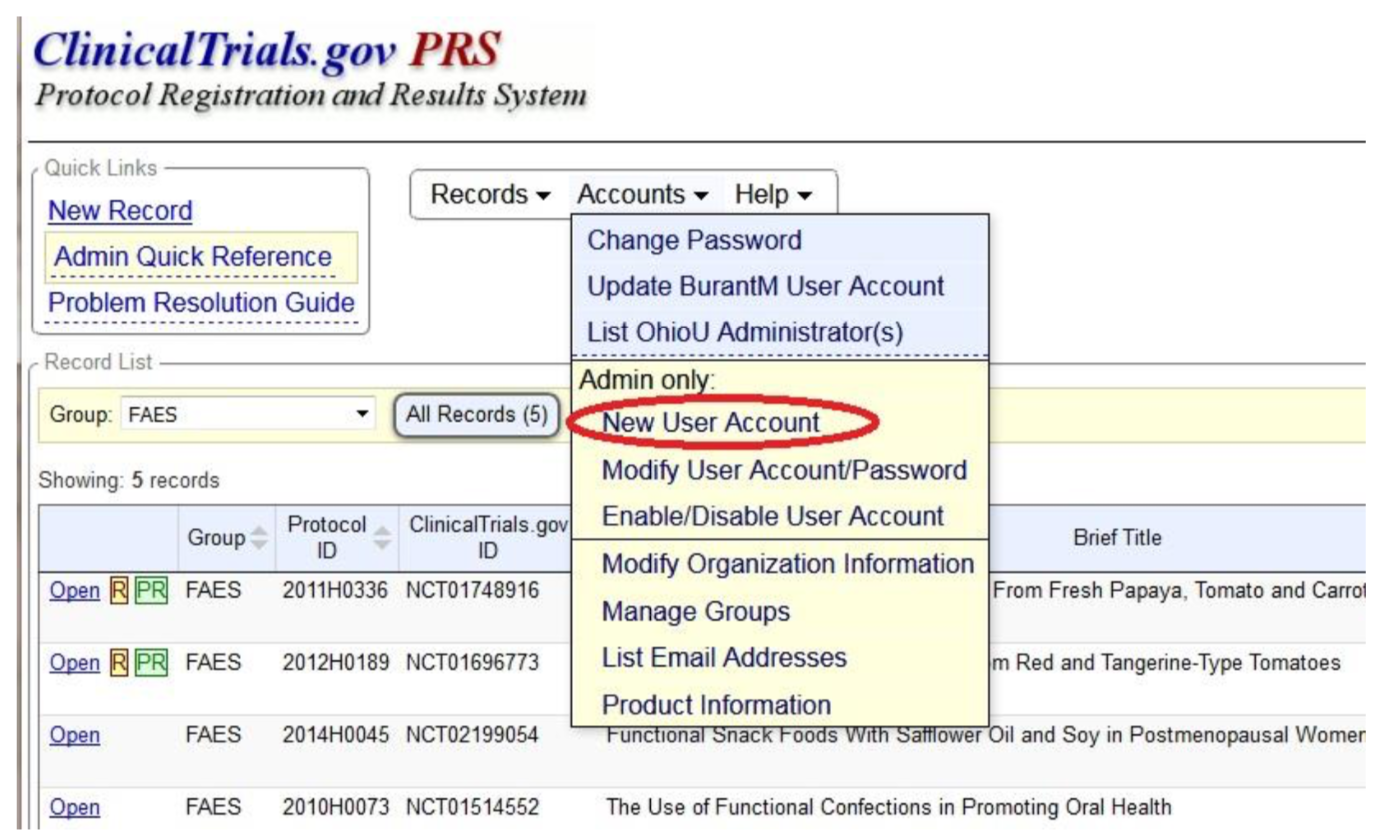
PRS Administrators create accounts for users in their college as needed, using the New User Account option from the Accounts menu on the top of the Clinicaltrials.gov PRS Home Page.
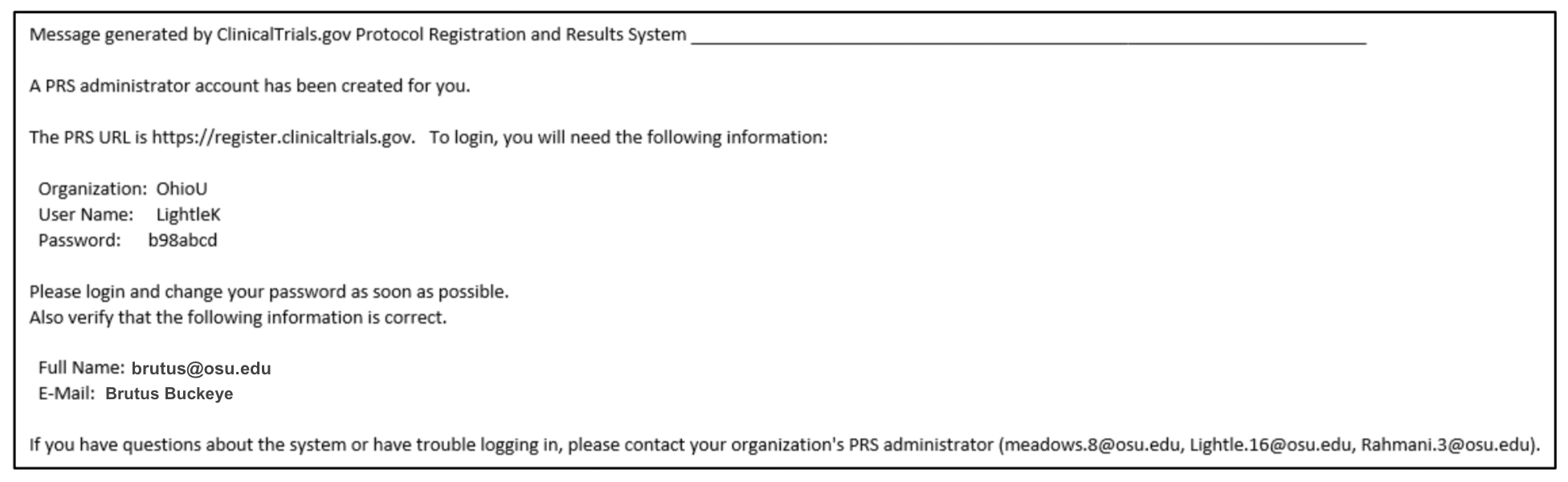
- The PRS automatically sends email with the PRS web address and login information to the user/administrator upon account creation.
- Administrators have full access to all records within the organization.
- Users create and modify their own records but normally cannot access other users' records.
- Study investigators are provided user accounts, not administrator accounts.
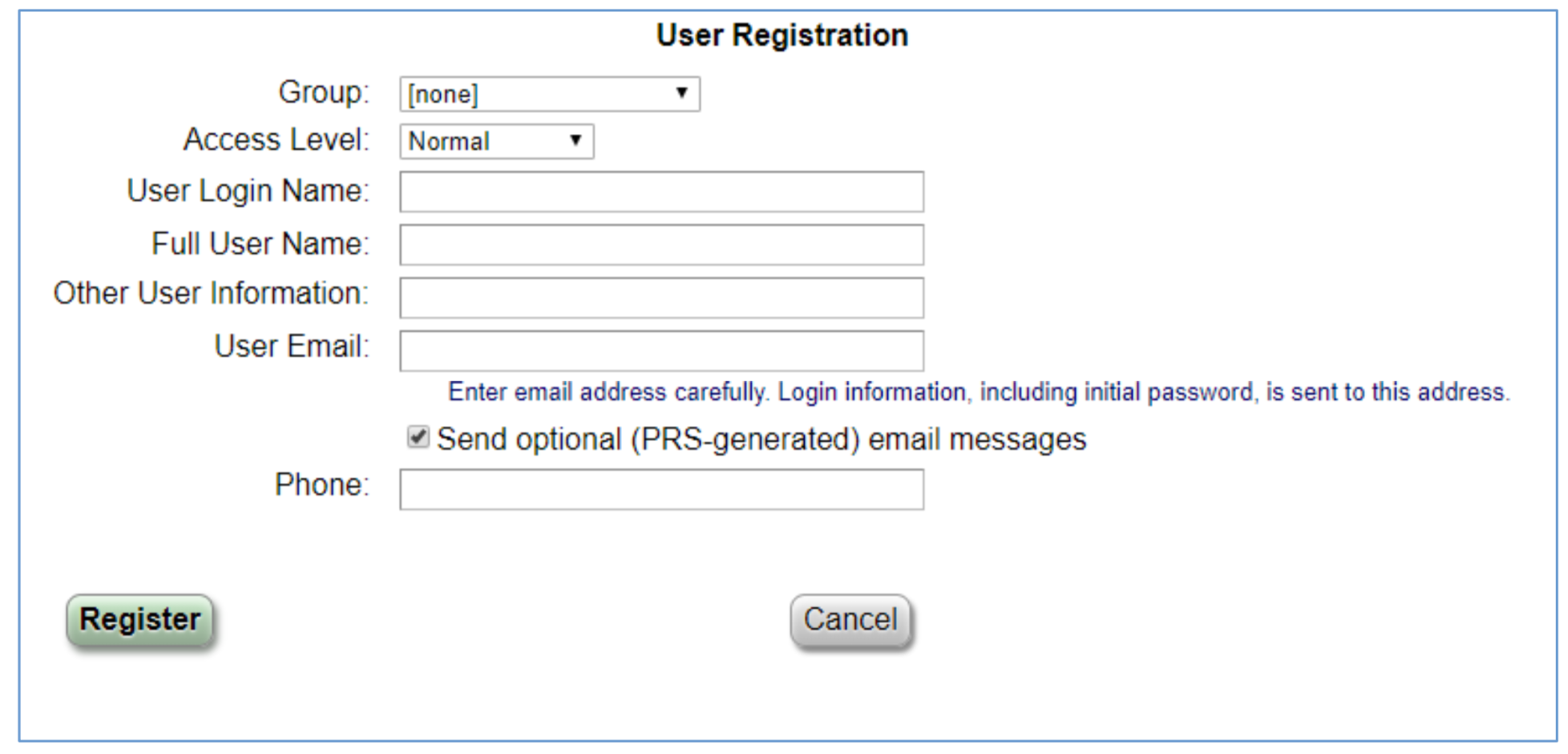
Best Practices for Filling in the Fields:
Group: Choose college from dropdown
Access Level: Normal
User Login Name: Ohio State name.#
Full User Name: First Last
Other User Information: Can put anything in this field, department is an option
User Email: Ohio State email
Send Optional (PRS-generated) email message: leave box checked
Phone: Ohio State Phone
Copy of email that new user receives from the PRS System:
Resources
Review, Approve and Release completed records
Whenever a user marks a new or modified record as Entry Completed, the PRS administrator is responsible for reviewing the record.
- Open the record and review all content, making corrections as needed.
- Follow instructions in the "Next Step" box on the "Record Summary" page to "Approve and Release" the record for PRS Review.
- Pay special attention to recruiting status and contact information when reviewing records, as the accuracy and timeliness of this information is extremely important to patients and health care providers.
- All records must be free of ERROR messages in order to be released.
- EXCEPTION: If the Investigator is designated as Responsible Party for a study, that individual has the authority (and responsibility) to "Approve and Release" the record even if not an administrator. In that case administrators can approve the record but cannot release it. More information on Responsible Party is accessible from the "Edit Sponsor/Collaborators" page.
Directions
Updating and Maintaining Records
Ensure that records are kept up to date
The administrator has overall responsibility to ensure that the organization's records are verified, updated and re-released as needed (at least yearly while a study is active).
- Email messages are automatically sent by the PRS to administrators for significant user actions, such as completing data entry for a record or modifying a previously released record.
- View the record specified in emails messages and check for problems.
- Use the Problems column on the Home page to identify important issues, such as records that were updated but never Released or records that are missing information required by the U.S. FDA Amendments Act (FDAAA 801).
- Work with the owner of each problem record as needed to address the issues, following instructions in the "Next Step" box on the Record Summary page to complete the process of updating the record.
- There are three basic reports available in PRS – "Home Page Report", "Public Site Report", and" Planning Report." Detailed information on how to access and best uses for all three can be found in the Introduction to CT.gov Reports document. Scheduling a time each month to review reports and follow up with record owners is a best practice.
Directions
Problems with Records and How to Resolve
Maintain user accounts
- Reset passwords via the "Accounts" menu on the Home page.
- Update email addresses or make other user account modifications via the "Accounts" menu on the Home page
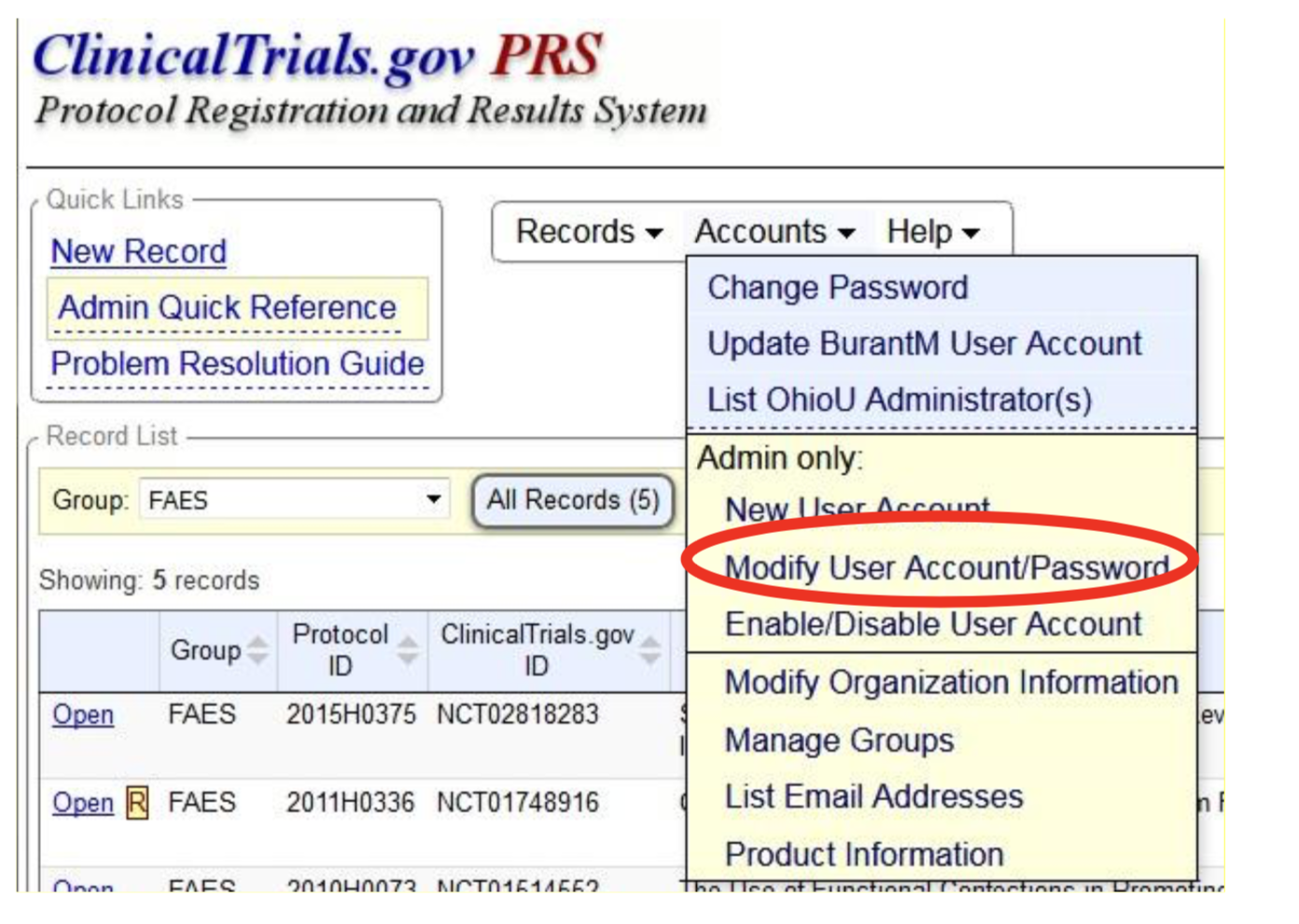
Update/Modify User Account Information
1. Click on "Modify User Account/Password" from the "Admin Only" section of the "Accounts"
2. Click on "Modify" next to the username for the account to be updated.
3. Enter changes on the "User Information" page.
4. Click "Save."
Change User Password
1. Click on "Modify User Account/Password" from the "Admin Only" section of the "Accounts" menu
2. Click on "Reset Password" next to the username for which the password is to be reset.
3. Enter and confirm a new password.
4. Click on "Reset Password." The new password will be emailed automatically to the user.
Directions
Update/Modify User Account Information
Disable user accounts
Disable a user's account using the "Enable/Disable User Account" option under the "Accounts" menu on the Home Page. This is often necessary when a user leaves the organization or no longer requires access to the PRS. Also, when users have multiple accounts.
Directions to disable/enable User Accounts
Maintain Access Lists for records
A record owner or PRS administrator can optionally grant access to a record, to one or more additional users within the same organization, using the "Edit Access List" link on the "Record Summary" page. Users granted access to a record in this manner have the same privileges as the record owner, except for modification of the "Access List."
Information for Investigators
- Objective
To ensure that the Principal Investigator (PI) and all research team members assisting in the conduct of clinical research are informed about their obligations and responsibilities as they pertain to Good Clinical Practices (GCP), the investigational plan, applicable regulations, guidances, and institutional policies. This Standard Operating Procedure (SOP) applies to the written procedures followed by all members of a clinical research team involved in the conduct of human subjects’ research at The Ohio State University Wexner Medical Center (OSUWMC), hereafter called the investigational site. These detailed instructions promote compliance in conducting clinical research.
SOP-20 describes the process for the registration and results reporting of clinical trials to ClinicalTrials.gov. Attachment templates include:
A: Creating a New Study Record for ClinicalTrials.gov
B: Maintaining Study Records on ClinicalTrials.gov - Responsibility
The College of Medicine Clinical Trials Management Organization (COM-CTMO) develops, implements, and maintains SOPs. The need to write a new or revise an existing SOP is based upon changes to federal regulations, guidelines, institutional policies, or procedures. These documents will be provided to departments and research teams conducting human subjects’ research. Departments or research teams may develop additional research SOPs or a Research Procedure Addendum (RPA) to expand on an existing SOP, however this need should be limited.
The PI is ultimately accountable for all clinical research activities and is responsible for the appropriate delegation of tasks to individuals with adequate training and education to perform such tasks. It is the responsibility of all members of the clinical research team involved in supervising, managing, or conducting study-related activities to follow the SOPs. The clinical research team may include but is not limited to the following members:
Research Team Members- Principal Investigator (PI)
- Clinical Research Coordinator (CRC)
- Sub-Investigator (Sub-I)
- Clinical Research Assistant (CRA)
- Clinical Research Manager (CRM)
- Other Research Staff as appropriate
- Clinical Research Specialist (CRS)
- Administrative and Support Staff
- Definitions (as they pertain to registering a trial on ClinicalTrials.gov)
- Applicable Clinical Trial (ACT): Registration is required for trials that meet the FDAAA 801 definition of an "Applicable Clinical Trial" and were either initiated after September 27, 2007, or initiated on or before that date and were still ongoing as of December 26, 2007. Trials that were ongoing as of September 27, 2007 and reached their completion date (see Primary Completion Date data element on ClinicalTrials.gov) before December 26, 2007 are excluded. "Applicable Clinical Trials" include the following:
- Trials of drugs and biologics: Controlled clinical investigations, other than phase 1 clinical trials that do not fall under FDAMA, of drugs or biological products subject to Food and Drug Administration (FDA) regulation.
- Trials of devices: 1) Controlled trials with health outcomes of devices subject to FDA regulation, other than small feasibility studies where the primary outcome measure relates to feasibility and not to health outcomes, and 2) Pediatric Postmarket Surveillance required by FDA.
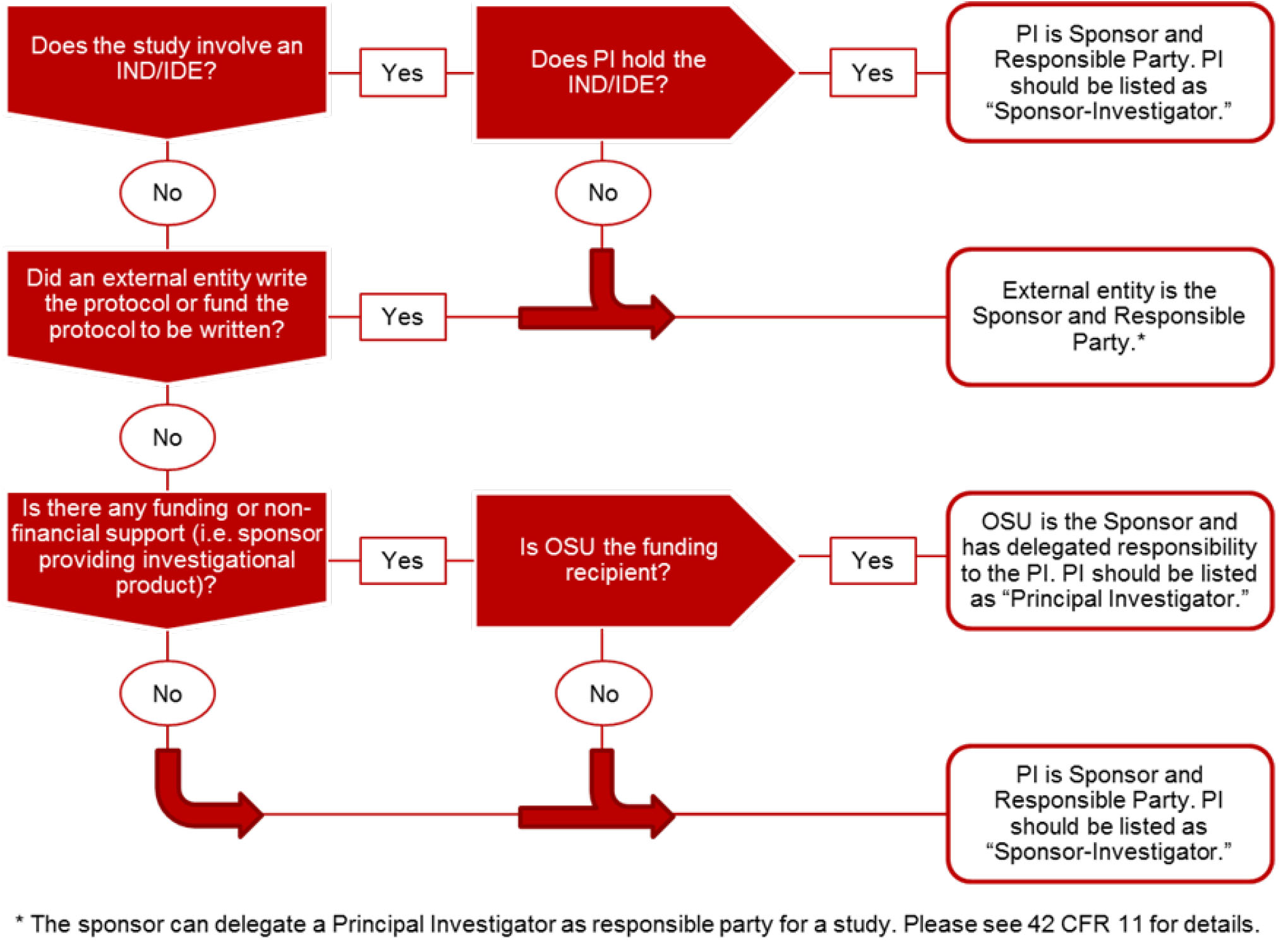
- Responsible Party: The individual with complete access to trial data and rights to publish. The Responsible Party for Investigator Initiated studies in the College of Medicine is the Principal Investigator. The Responsible Party may designate individuals to help complete the ClinicalTrials.gov record, however, the final responsibility of review and approval lies with the Responsible Party. The Responsible Party has the sole authority to release a record.
- Record Owner: This individual can be the Principal Investigator or a designated research team member that is responsible for updating the ClinicalTrials.gov record and ensuring that it is updated in a timely manner. The owner must maintain communication so that the protocol record is released by the PI (Responsible Party) in the required time frame.
- International Committee of Medical Journal Editors (ICMJE): A small group of general medical journal editors and representatives of selected related organizations working together to improve the quality of medical science and its reporting.
- Protocol Registration and Results System (PRS): Website for entering and updating ClinicalTrials.gov records. https://register.clinicaltrials.gov/
- Applicable Clinical Trial (ACT): Registration is required for trials that meet the FDAAA 801 definition of an "Applicable Clinical Trial" and were either initiated after September 27, 2007, or initiated on or before that date and were still ongoing as of December 26, 2007. Trials that were ongoing as of September 27, 2007 and reached their completion date (see Primary Completion Date data element on ClinicalTrials.gov) before December 26, 2007 are excluded. "Applicable Clinical Trials" include the following:
- Procedures
- Determining Study Registration Requirements:
The PI is responsible for determining if the study is an ACT, thus requiring registration and results information on ClinicalTrials.gov. To determine if a study is an applicable clinical trial per 42 CFR 11, please use the checklist available on the ClinicalTrials.gov website
(https://prsinfo.clinicaltrials.gov/ACT_Checklist.pdf).
In addition to federal requirements (42 CFR 11) and the NIH Policy, journals (i.e., ICMJE), funding sources, and insurance companies (i.e., Medicare/Medicaid) may require registration and submission of results information.
If the study was approved by the Cancer IRB, the PI will be instructed to work with appropriately delegated ClinicalTrials.gov Administrator within the Comprehensive Cancer Center to ensure research studies are registered appropriately.
Studies with approved consent language stating the study will be registered to ClinicalTrials.gov must register their study to ClinicalTrials.gov. If it is determined that the study does not require registration per regulations and the PI decides not to register the study for publication or other purposes, then an amendment removing the ClinicalTrials.gov language must be approved by the IRB and consented subjects
must be notified.
Studies with human subjects that are not required per regulations or publication requirements may still be registered to ClinicalTrials.gov per the PI’s discretion. - Determining Responsible Party
Please use the flow diagram and links below, as well as the NIH’s flow sheet and information on the ClinicalTrials.gov website to determine who the Responsible Party is and what option should be selected. For studies initiated and written by an investigator at OSU, the PI of the study should be listed as the Responsible Party, whether listed as “Principal Investigator” or “Sponsor-Investigator.” The Responsible Party has the sole authority to approve and release the record; all records must be reviewed and released by the Principal Investigator. In the event that the PI leaves OSU, please contact the administrators to determine who should become the new Responsible Party.
http://grants.nih.gov/ClinicalTrials_fdaaa/Responsible_Party.htm
http://prsinfo.clinicaltrials.gov/ElaborationsOnDefinitions.pdf
- Obtain an Account
The Ohio State University has separate ClinicalTrials.gov administrators for cancer and non-cancer clinical research studies.- Requests for a ClinicalTrials.gov account can be made by contacting the appropriate organizational ClinicalTrials.gov administrator.
- The requestor must provide full name, preferred institutional e-mail address, IRB number and title of the protocol. If the requestor is not the PI, an e-mail from the PI to the ClinicalTrials.gov administrator is required to create or change access. Please note that the PI of the study will need to have a ClinicalTrials.gov account in order to approve and release the record. You may request that at the same time if the PI does not have an account yet.
- Once an account is created you will be notified by the administrator and will receive an e-mail from the Protocol Registering System (PRS) to modify your password.
- Instructions to create, edit, approve and release a study record will be provided to the PI and record owner.
- Confirm Responsible Party (Refer to section B for the definition of “responsible party”): Use the following guidance flow diagram to determine if who is the responsible party:
https://grants.nih.gov/ClinicalTrials_fdaaa/docs/registration_flow_chart.pdf .
- Create, Update and Maintain Study Records
Instructions for creating, editing, approving, and releasing a study record are available on the main ClinicalTrials.gov website.- Creating and updating submissions: For new records please refer to Attachment A: Creating a New Study Record for ClinicalTrials.gov.
- Maintaining record: please refer to Attachment B: Maintaining Study Records on ClinicalTrials.gov.
- Records release: All records must be reviewed and released by the PI.
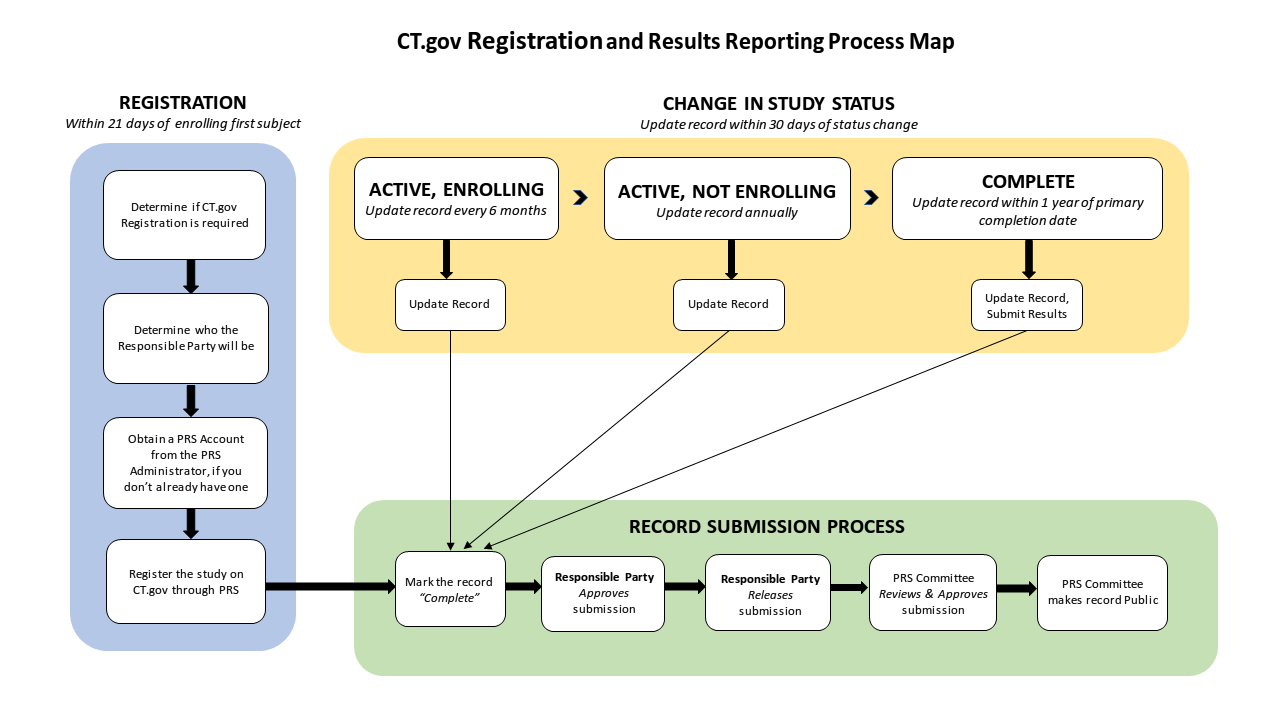
- Timeline Requirements
Event Timeline requirements Notes Registration No later than 21 days after the first subject is enrolled
ICMJE requires that registration is complete prior to first subject enrollment.A study is considered registered once the responsible party releases the record to PRS for review. Actively enrolling studies Update/verification every 6 months The record must be verified even if no changes need to be made. Studies closed to enrollment or pending results Update/verification annually The record must be verified even if no changes need to be made. Change in study status Within 30 days of status change
ResultsResults submission No later than 1 year after the primary completion date Delayed submission of results is permitted in certain circumstances. See 42 CFR 11.44 for details. - Protocol Record Management
- The Responsible Party is ultimately responsible for ensuring the studies are registered with ClinicalTrials.gov and updated appropriately at required intervals and released to the public database. Refer to Section D above for timeline requirements for updates.
- The PI and protocol Record Owner will be contacted by the appropriate ClinicalTrials.gov administrator if their protocol record is delinquent and needs to be updated.
- If the protocol record remains delinquent two weeks after the first notice, a second notification will go out to the PI, protocol Record Owner and department chair.
- If the protocol record remains delinquent one month after the initial contact without acceptable activity/progress, the COMOR will arrange for alternative management of the account by another party. The cost of these services will be billed to the department.
- Records that are entered into the ClinicalTrials.gov database, but are not released, will get one notification and if left incomplete at 30 days post notification, the record will be deleted from the database.
- Please note that records cannot be deleted once they have been issued an NCT number, even after the study has been completed. There are limited circumstances when a record can be removed from the public site – please contact the administrator for assistance.
- Transferring a Record:
- If the Record Owner or PI is leaving the Institution they should inform their ClinicalTrials.gov Administrator to ensure the record is appropriately monitored or transferred. The Record Owner or PI can either be reassigned to another Record Owner or PI within the university, or the record can be transferred to a new institution. The ClinicalTrials.gov organizational name for the institution where the PI is transferring should be provided. If this information is unavailable at the time, contact information should be provided to facilitate transfer.
- If the PI (Responsible Party) is moving studies from another institution please contact your designated ClinicalTrials.gov administrator to help facilitate the record transfer.
- Penalties
- Under 42 CFR 11, civil and monetary penalties exist for noncompliance. Monetary penalties can be up to 10,000 US dollars a day.
- Grant funding can be withheld until the required clinical trial information has been submitted.
- Journals can refuse to publish data from records that are noncompliant.
- Noncompliance with OSU policies, 42 CFR 11, and other requirements could result in corrective actions that may include reporting of noncompliance to the IRB.
- Determining Study Registration Requirements:
- Applicable Regulations, Guidances and Policies
Regulation/ Guidance/Policy Title 42CFR11 Final Rule NIH Policy on Clinical Trial Registration What NIH Grantees Need to Know About FDAAA Food and Drug Modernization Act Section 113 Information Program on Clinical Trials for Serious Life-Threatening Diseases Food and Drug Administration Amendments Act (FDAAA) Section 801 Expanded Clinical Trial Registry Data Bank Declaration of Helsinki Ethical Principles for Medical Research Involving Human Subjects World Health Organization International Clinical Trials Registry Platform
(ICTRP)International Committee of Medical Journal Editors Clinical Trial Registration Food and Drug Administration Amendments Act (FDAAA) Section 801 Elaboration of Definitions of Responsible Party & Applicable Clinical Trial ORRP Guidance Clinical Trials Registration Office of Responsible Research Practices guidance on Clinical Trial Registration
Additional Resources